The ideal gas law is 
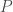
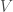



Recently, I’ve come to appreciate the ideal gas law as a very good illustrative example of what statistical mechanics is capable of. In the eighteenth and nineteenth (during the industrial revolution), the field of thermodynamics arose in pursuit of answers to how much energy can be extracted from systems. At the time, the notion of temperature was hotly debated, with one ostensibly reasonable theory being that the flow of a fluid, called “caloric,” facilitated heat transfer. It should be appreciated just how many important results could be derived by our predecessors without precise knowledge of the microscopic physics involved.
Statistical mechanics arises from a desire to understand how thermodynamics arises from microscopic physics, either classical or quantum. While classical thermodynamics is able to achieve a great deal, it is philosophically difficult to reconcile the microscopic and macroscopic worlds without some statistical framework that can translate the former to the latter. Moreover, it allows us to determine the thermodynamic behavior of new systems that are hard to reason about a priori—how, for example, should we expect the stars in a star cluster to behave? What about a complicated, novel quantum field theory?
In this article, I will present a number of different derivations of the ideal gas law within the framework of statistical mechanics. In the process, I hope to motivate the different ensembles of statistical mechanics (which ultimately just encode the behavior of possibly large systems for which only a small amount of bulk information is known). In the process, I hope that an overview of these derivations will be a useful introduction for current physics students (or other interested people) to a notoriously (but arguably needlessly) opaque field.
A hand-wavy, “kinetic” derivation
Before we proceed with derivations from statistical mechanics, I think that there is utility in understanding at a heuristic and intuitive level what is being stated by the ideal gas law. An ideal gas is a simplified model which models a gas as a collection of very small, collisionless particles which exert forces on the sides of a container (e.g., a box) whenever they collide with them. The pressure is a reflection of this force, and is (by definition) force per unit area. This can go up if (1) the number of particles per unit volume is increased (so that more particles are around to hit the sides of the box), or if (2) the typical velocities (read: temperature) of the particles are increased (so that each collision of a particle with the sides of the box imparts more momentum on average).
We can start by saying that the pressure can also be thought of as the normal momentum flux, or the amount of momentum perpendicular to the sides of the box which moves through a surface per unit area (this is equivalent to the force per unit area, since force is momentum per time). We can write

For particles with mass 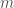

At a temperature 


or

In fact, this approximate expression is actually exact, and kinetic theory can be used to show that the numerical factor that ultimately ends up in front is 
How to think about statistical mechanics
Statistical mechanics (as mentioned above) is a formalism for transforming microscopic physical laws into thermodynamics. Classical thermodynamics, in turn, is based on the determination of so-called thermodynamic potentials, of which the most famous is energy 


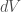

where 
Another is the entropy 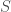
The other “thermodynamic potentials” are obtained by performing a mathematical operation called a “Legendre transform,” whose interpretation is very neat and summarized well here. For example, we can define the Helmholtz free energy as 


We see in the differential that the effect of subtracting 



The differential quantities which naturally appear in the above expressions should be interpreted as the “natural” quantities of the potential. While that seems a bit hand-wavy, I mean that, for example, if you were doing an experiment where you had control over the temperature, pressure, and particle number (as chemists often do), it would probably be most useful for you to reason about your system through the Gibbs free energy, where 

Moreover (and relatedly), the knowledge of any of these potentials as a function of their natural variables will encode all the macroscopic thermodynamical quantities. For example, if we had knowledge of the energy function 

where subscripts indicate that a partial derivative is being taken with the subscripted variables fixed (for example, 




Hence, by taking derivatives of our known energy function 

whereby 

where 
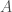

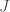
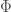

As a fun aside, it turns out that performing a Legendre transform with respect to all particles in a system is “not allowed” in the sense that it will actually cause the quantity to vanish entirely:

This result is called the Gibbs-Duhem relation.
Fantastic ensembles and where to find them
Heuristically, most, if not all, equilibrium statistical mechanics problems go as follows: we count the number of microscopic states (“microstates”) which are compatible with a given macroscopic state, and then straightforwardly translate that result into a thermodynamic potential which is most natural to that system. Upon obtaining that thermodynamic potential, we can then use rules from classical thermodynamics to find out any thermodynamical quantity about our system that we want.
A fundamental assumption of statistical mechanics is the principle of indifference, a (sometimes debated) postulate that two microscopic configurations of a system which have the same total energy will be equally probable. A shuffled deck of cards is unlikely to be in order, not because any given shuffled deck is more likely than an ordered deck, but rather because there are many more ways for the shuffled deck to be out of order. From this perspective, it is a little bit clearer why solving problems in this field almost always involves counting states, sometimes weighted by some quantity to account for coupling to a heat or particle bath.
Statistical mechanics reasons about ensembles, which can be thought of as probability distributions over a given system where certain macroscopic quantities are known (possibly including, for example, the temperature). Alternatively, they can be thought of as a large collection of macroscopically (but not necessarily microscopically) identical systems. The sense in which the systems are macroscopically identical depends on which type of ensemble is being used (i.e., which macroscopic quantities are shared in common between the systems which makes them “macroscopically identical”).
However, an important fact must be stated: for systems with very many particles, the ensembles ought to agree. On a macroscopic level, an isolated system ought to behave identically to the same system which is in contact with a heat bath of the same temperature. Even though, strictly speaking, the latter is coupled to another system and the former is not, the lack of heat flow means that the systems should be macroscopically indistinguishable. The same goes for a particle bath with the same chemical potential as the system.
I will briefly summarize below the three most common statistical ensembles which are introduced in usual courses on statistical mechanics (although this is not an exhaustive list of all ensembles which could exist). There are some very common tricks to doing problems in all of the ensembles, some of which I will also describe below. The ultimate hope is that the way to utilize those tricks will become clear in the concrete example of an ideal gas.
Microcanonical Ensemble
In the microcanonical ensemble, we know a system’s total energy 
In such problems, we will seek to count the total number of states 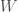
![[E,E+\delta E]](https://s0.wp.com/latex.php?latex=%5BE%2CE%2B%5Cdelta+E%5D&bg=ffffff&fg=404040&s=0&c=20201002)

where

In the case of continuous states, for example of 
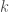

where 
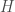





\noindent where 

After finding
and from here can use classical thermodynamics to find out everything else.
Canonical Ensemble
In the canonical ensemble, we again know the system’s volume 

Again, we are required to count the number of states, but this time with higher energy states being exponentially down-weighted (see, e.g., here for a satisfying derivation from Lagrange multipliers). In the discrete case, we have
This time, the sum 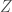



Note that, if the system comprises many particles with identical behaviors whose energies sum together, i.e., 


where 
Because the natural variables for this ensemble are now 
Again, after obtaining 
To reiterate, even though in this case the total energy of the system
Grand Canonical Ensemble
In the grand canonical ensemble, we know the average energy per particle 
Counting the states this time requires computation of the so-called grand canonical partition function 
where the Boltzmann factors 

In both cases, we can factor 


Hence, if the canonical problem has already been solved, it is often not too difficult at least in principle to extend it to the grand canonical case.
The natural variables 
in a similar manner to
Once again, even though a particle bath is only involved here, in realistically large systems the total particle number
Deriving the ideal gas law in the classical (continuous) case
Since all the ensembles should yield the same results in macroscopically large systems, all three ensembles described above can be used to derive the ideal gas law (although not necessarily with the same ease).
Furthermore, we can approach this problem both from classical (continuous) physics (which involves integrating over phase space) or take the high temperature limit of the quantum (discrete) case (which involves summing over states). Both of these approaches should, again, give the same answer as they should converge to the same physics at the regimes that we care about.
Microcanonical Ensemble: Classical Case
We will seek to find the number of states
Since the positions play no part in the energy of a particle within an ideal gas, the integrals 


We may be tempted to evaluate the integral above by hand, but that would probably result in many empty boxes of chalk filled with tears.
Instead, we can find the number of states geometrically, by noting that the Hamiltonian (energy) for such a system is simply the sum of the kinetic energies:

The hypersurface 

which looks awfully similar to the formula for a sphere (cf. 




Nevertheless, we trust that mathematicians have taken care of this problem, and indeed a quick look at Wikipedia reveals that a 

where 


Then we can find

This can then be translated into an entropy which, using the rules for logarithms, cleans up immensely:

where 


Then we can write

We can then find the pressure by identifying

which yields the ideal gas law:
Canonical Ensemble: Classical Case
We saw that one difficulty with the microcanonical ensemble was that the integrals were too hard to do algebraically (at least, if the goal is to still want to be alive afterwards). The fundamental issue is that, when we fix the total energy of the system
In contrast, if we take the canonical approach of only fixing the average particle energy
We can compute the partition function which is analogous to state counting once again:
Again, since the energy of the gas does not depend on the location of the particles but only their momenta, we can quickly evaluate the position integrals:

where we have substituted in the Hamiltonian for an ideal gas. Notably, the exponential can be broken up as follows:

We see that each factor in the integrand only depends on one component of one particle’s momentum, so we can pull each factor out of all the integrals that don’t depend on it, and we will obtain

We can recognize this as

If we had been clever from the start, we could have recognized that, in fact, the total partition function
where

Anyway, we can now translate to the macroscopic picture by deriving the Helmholtz free energy:

We can then write

Aiming for the pressure once again, we can identify

which, once again, leads to the ideal gas law:
Grand Canonical Ensemble: Classical Case
If we insisted in allowing the particle number to vary, we could, in fact, verify that we would achieve the same ideal gas law even if, say, the system were coupled to a large particle bath. We can work in the grand canonical ensemble, where now rather than knowing
We now start by performing the relevant “state counting,” which in this case involves deriving the grand canonical partition function:
We recognize that the integral has a factor 

Before doing literally anything else (even the trivial volume integrals), we can step back and recognize that the integral within the sum over

Noting that everything in the summand is exponentiated to the

we recognize that the grand canonical partition function is, in fact, a geometric series:

We can then relate this to the “natural potential,” in this case the grand potential

We note that

We can again identify

This is ostensibly a very ugly formula, for which we are required to eliminate

We see that the mess

appears in our expression for

where, again, we find the ideal gas law.
Deriving the ideal gas law in the quantum (discrete) case
Quantum mechanics devolves to classical mechanics in the macroscopic limit—Ehrenfest’s theorem is that the expectation values of quantum mechanics will follow the usual classical equations of motion. Thus, we should not be particularly surprised that we can also derive the ideal gas law from a quantum mechanical perspective, with states now being discrete rather than part of a continuous phase space.
However, curiously, from a quantum mechanical perspective, the usual Boltzmann distribution is actually only a high temperature limit of the Fermi-Dirac or Bose-Einstein distributions, and does not arise on its own from any natural-looking partition function. In the Fermi-Dirac distribution (which describes fermions), two particles cannot share the same state. In the Bose-Einstein distribution (which describes bosons), they assuredly can (and, compared to the classical picture, are more zealous to do so particularly at low temperatures).
In the following derivations, I will be considering bosons, since counting states while also excluding those where multiple particles share a given state is somewhat more complicated. I will only perform the derivation up to the point where the mathematics becomes identical to the classical case, which must occur at some point for the physics that occurs to be identical.
This section will use some basic facts from quantum mechanics, in particular that the energy of a particle in an infinite square well with dimensions 



where the wavenumbers 





where 
Microcanonical Ensemble: Quantum Case
We would like to count all of the states where this energy is equal to the numerical value
As in the continuous case, we may instead consider all the states with energy

where
The total energy of the system can be written as

which can again be rearranged into the equation for a

In the single particle 






where the factor of 


which is exactly the same as the classical result. The rest of the analysis is identical to the classical microcanonical derivation, which can be found above.
One important note is that, in this case, the phase space volume of a state being 
Canonical Ensemble: Quantum Case
In the canonical ensemble, we would like to compute the partition function

where

We see that the sum in

The summands are Gaussians in 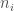



which evaluates to

This can be rewritten as

and the steps that follow are identical to the classical case.
Grand Canonical Ensemble: Quantum Case
In the grand canonical ensemble, we use the trick that we can decompose the grand canonical partition function 

However, because we have already found that the quantum mechanical canonical partition function
Summary
Equilibrium statistical mechanics comprises a set of tools for converting microscopic physics into macroscopic physics, in the absence of most of the information which describes the state of the system. When I first learned statistical mechanics, these tools seemed to be very ad hoc and disjoint from each other, but they fit together into an elegant, (debatably) coherent framework. When I now think about statistical mechanics, I have the following table in mind:
| Ensemble | Counting states (microscopic) | Potential (macroscopic) |
| Microcanonical |  | Entropy ( ) ) |
| Canonical |  | Helmholtz free energy ( ) ) |
| Grand Canonical |  | Grand potential ( ) ) |
When keeping in mind that the ultimate purpose here is to try to convert microscopic physics into macroscopic physics, we can learn insightful facts about the behavior of large systems, such as the ideal gas law, which can be found using any number of the tools presented in a typical undergraduate statistical mechanics course.
All roads lead to Rome.
